Table Of Content
Subsequently, this feature vector is decoded back into the corresponding molecular string, using a CLM based on an RNN-LSTM32,76 architecture for the molecule generation process. The study also included the evaluation of novelty, synthesizability, and predicted bioactivity for the generated molecules. These criteria were essential in assessing the practicality and potential value of the designed compounds. To quantify molecular novelty, a rule-based algorithm was utilized, which captured both, scaffold and structural novelty. This algorithm, described in Equations (9)–(12), offers a quantitative measure of the uniqueness of each molecule in terms of its chemical structure.
Pharmaceutical Sciences (PSCI)
Lately, Daniel Koshland suggested that both molecules do undergo conformational changes during interaction and adopt the most suitable conformation in order to connect each other. This hypothesis has been proven many times by X-ray structures and in silico simulations and now it is known that ligands indeed change their conformations during the interaction and adopt conformations that optimally fit the contact surfaces. Quantitative structure–activity relationshipmodels (QSAR models) are regression or classification models used in the chemical and biological sciences and engineering.
Pre-clinical drug discovery
A Pharmacist's duties include dispensing medication, compounding medicine, giving advice on health, and providing immunizations. It should be noted that not all programs listed below offer the certification or licensing to become a Pharmacist, but only provide the necessary educational background, or may be geared towards professionals already in the field. The umbrella structure allows students to attend courses and seminars together, and rotate through laboratories across programs during their first year. This fosters interdisciplinary crosstalk among students and faculty, helping students find an ideal laboratory and faculty mentor as well as a specialized track of study they want to pursue. Upon successful completion of the first year, students will select an area of study from one of the three listed below, in which they will earn the Doctor of Philosophy degree following successful completion of the PhD Program.
Receptor-Based Design¶
To build a model that can implement the sequence-to-drug concept, we developed TransformerCPI2.0 based on our previous work20 and its framework is shown in Fig. As pointed out in our previous work, there is a common hidden ligand bias issue in existing CPI datasets20. Therefore, we ensured that each compound in our dataset exists in positive class and negative class, but pairs with different proteins. Because the compounds with positive and negative labels are exactly the same, ligand bias is greatly reduced in our dataset.
3D molecular generative framework for interaction-guided drug design - Nature.com
3D molecular generative framework for interaction-guided drug design.
Posted: Wed, 27 Mar 2024 07:00:00 GMT [source]
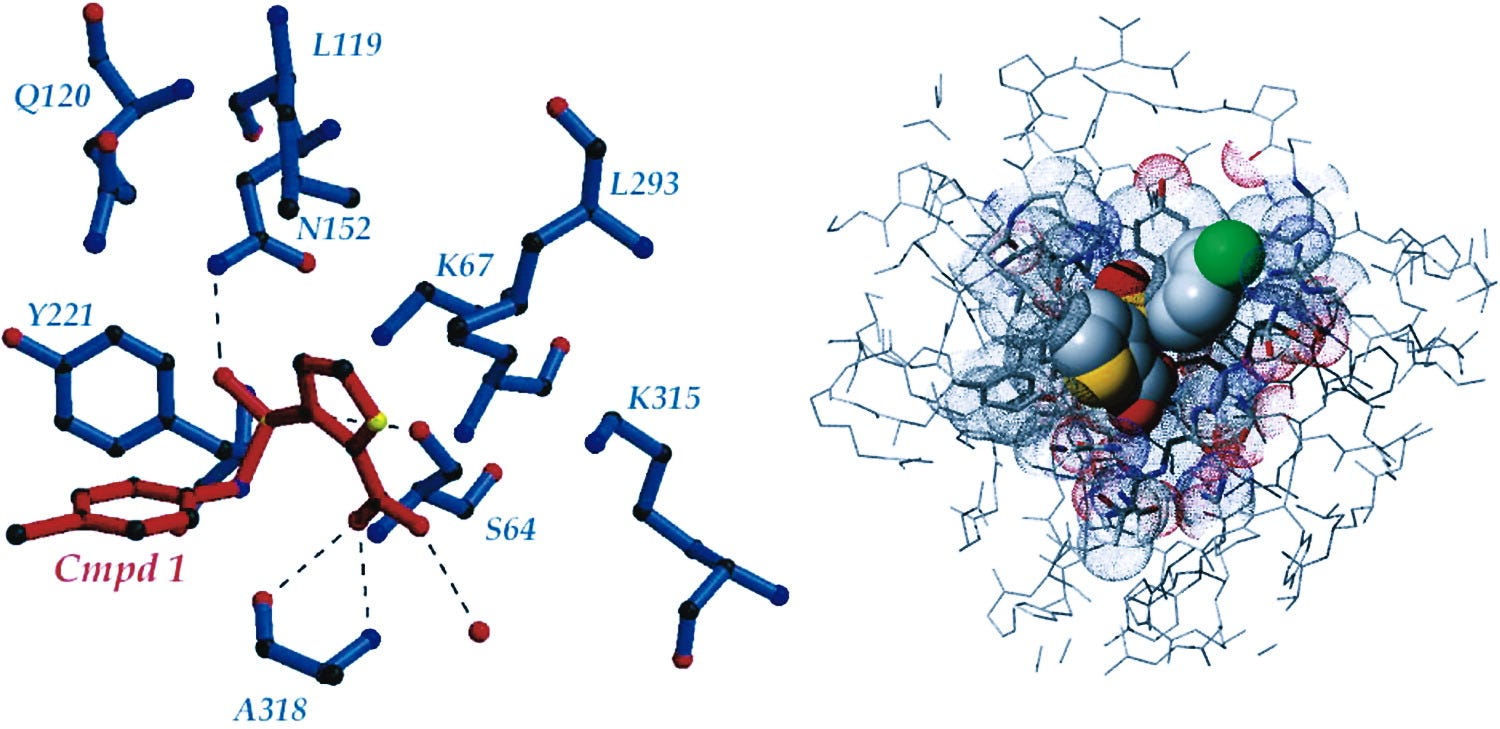
An amino acid site whose contribution to compound–protein binding is large can be revealed quantitatively by the relative activity change score ΔR. Finally, we used the activity change score ΔS to plot a heatmap to study the concrete pattern of drug resistance mutations and the relative activity change score ΔR to rank the most important sites for compound protein binding. The conventional structure-based drug design pipeline is a complex, human-engineered process with multiple independently optimized steps. However, the multistep operation is error-prone due to factors such as inaccurate protein structures, the multiplicity and dynamics of binding pockets, incorrect pocket definition, inappropriate selection of the scoring function, etc.
Coursework emphasizes molecular pharmacology and the interaction of drugs with cell physiology. Research areas are laboratory-based and include drug design and development, receptor pharmacology, pharmacodynamics, medicinal chemistry, cancer biology and pharmacology, immunology, metabolism and biochemistry, molecular- and neuro-pharmacology, and oxidant and environmental toxicology. The MPTX PhD program and the PSCI PhD program are both administered under the umbrella of the Department of Pharmacology and Pharmaceutical Sciences. To confirm the feasibility of sequence-to-drug concept, we compared TransformerCPI2.0 with conventional structure-based drug design approaches to test its ability to screen active molecules from compound libraries.
Life is based on molecular systems that are organized and regulated by precise recognition and discrimination process. Insights into biological systems show us that all the underlying molecular machinery operates in three-dimension. They report for the first time that dutasteride influences the protein expression profiling in the epididymal luminal fluids of rats, and this result provides some new epididymal targets for male contraception and infertility therapy. The study results are presented in the article “Identification of New Epididymal Luminal Fluid Proteins Involved in Sperm Maturation in Infertile Rats Treated by Dutasteride Using iTRAQ” [12].
Molecules generated with DRAGONFLY potently and selectively activate PPARγ
The binding parameters of the compound iRNF to RNF130 were measured with a MicroCal PEAQ-ITC calorimeter. The SPR binding assay was performed using a Biacore T200 instrument (GE Healthcare). The purified RNF130 protein was covalently immobilized onto a CM5 sensor chip (Cytiva) by a standard amine-coupling procedure in 10 mM sodium acetate (pH 4.5) with running buffer HBS (50 mM HEPES pH 7.4, 150 mM NaCl). IRNF was serially diluted and injected onto the sensor chip at a flow rate of 30 μL/min for 120 s (contact phase), followed by 120 s of buffer flow (dissociation phase).
Meta-learning for transformer-based prediction of potent compounds
After the ETH researchers had produced these molecules in the lab, colleagues at Roche subjected them to a variety of tests. These showed that the new substances are indeed stable and non-toxic right from the start. The new method builds on the decades-long efforts of chemists to elucidate the three-dimensional structure of proteins and to use computers to search for suitable potential drug molecules.
The ASO prevented the splicing factor from binding to the TEAD mRNA, so it couldn’t ultimately splice it out. We got pretty much only inactive TEAD when we treated MASH mice with the ASO,” said Olefsky. “All the molecules in the pathway were known, but no one knew if or how they interacted. We put the pathway together, showing each step in this intracellular signaling module. The clinical message is this ASO, which can actually block liver fibrosis,” said Jerrold Olefsky, a professor of medicine and assistant vice chancellor for integrative research at UC San Diego Health Sciences, and senior author on the paper.
Affecting the functions of target macromolecule could change the etiology of the disease, the pathophysiology or simply improve the symptoms. Of the 20,000 protein-coding genes in the human genome, about have been estimated to be part of the so-called druggable genome or druggable proteins [27]. Oprea et al. [28] divided the whole proteome into four groups according to the target development level, i.e., how much we know about a given protein (Figure 2).
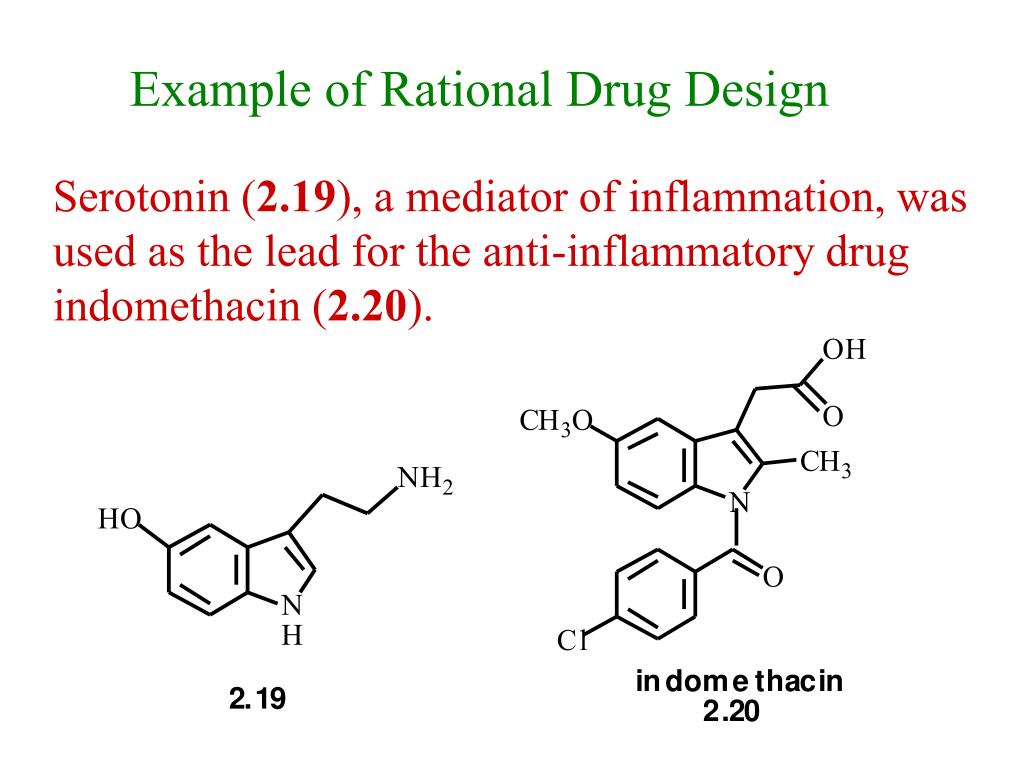
While many quantitative structure activity relationship analyses involve the interactions of a family of molecules with an enzyme or receptor binding site, QSAR can also be used to study the interactions between the structural domains of proteins. Protein-protein interactions can be quantitatively analyzed for structural variations resulted from site-directed mutagenesis. The current trend in the drug design is to develop new clinically effective agents through the structural modification of lead nucleus. The lead is a prototype compound that has the desired biological or pharmacological activity but may have many undesirable characteristics, like high toxicity, other biological activity, insolubility or metabolism problems.
First, we examined the generalization ability across proteins and chemical space. Second, we designed drug resistance mutation analysis and substitution effect analysis to interpret our model whether it learns knowledge as expected. Third, we applied a sequence-to-drug concept to screen new hits for challenging targets and novel targets without 3D structures, and conducted drug repurposing task.
In ccRCC cells, SPOP is overexpressed and misallocated in the cytoplasm, inducing proliferation and promoting renal tumorigenesis47. Two substrates of SPOP are phosphatase and tensin homolog (PTEN) and dual specificity phosphatase 7 (DUSP7)47. PTEN acts as a negative regulator of phosphoinositide 3-kinase/AKT pathway, and DUSP7 dephosphorylates extracellular signal-regulated kinase (ERK)48. The accumulation of cytoplasmic SPOP in ccRCC cells decreases cellular PTEN and DUSP7 by mediating the degradation of these two cytoplasmic proteins, leading to an increase in phosphorylated AKT and ERK and promoting ccRCC cell proliferation47. The metabolism of opioids, also considering their low clinical dosage, always attracted the attention of many investigators [75].








No comments:
Post a Comment